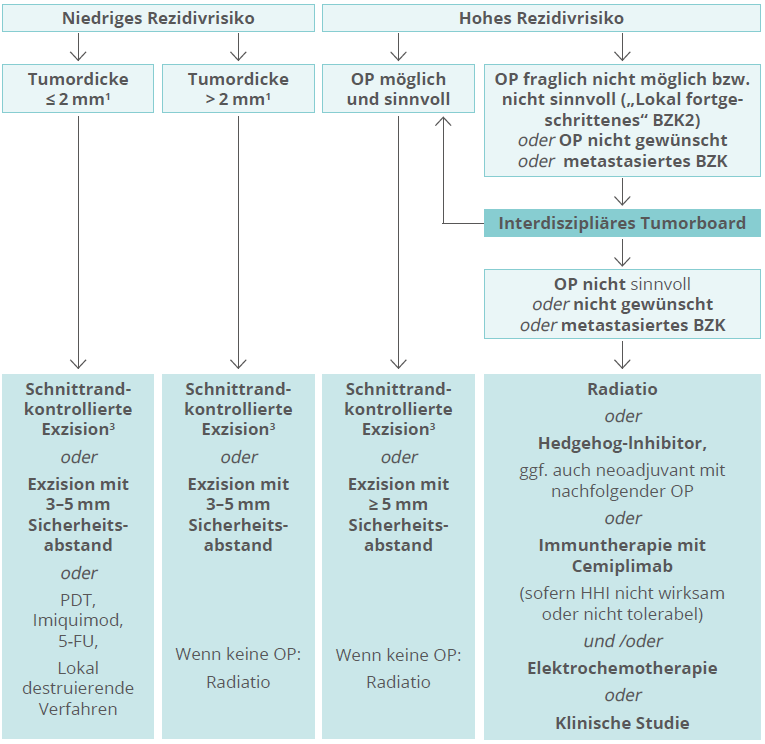
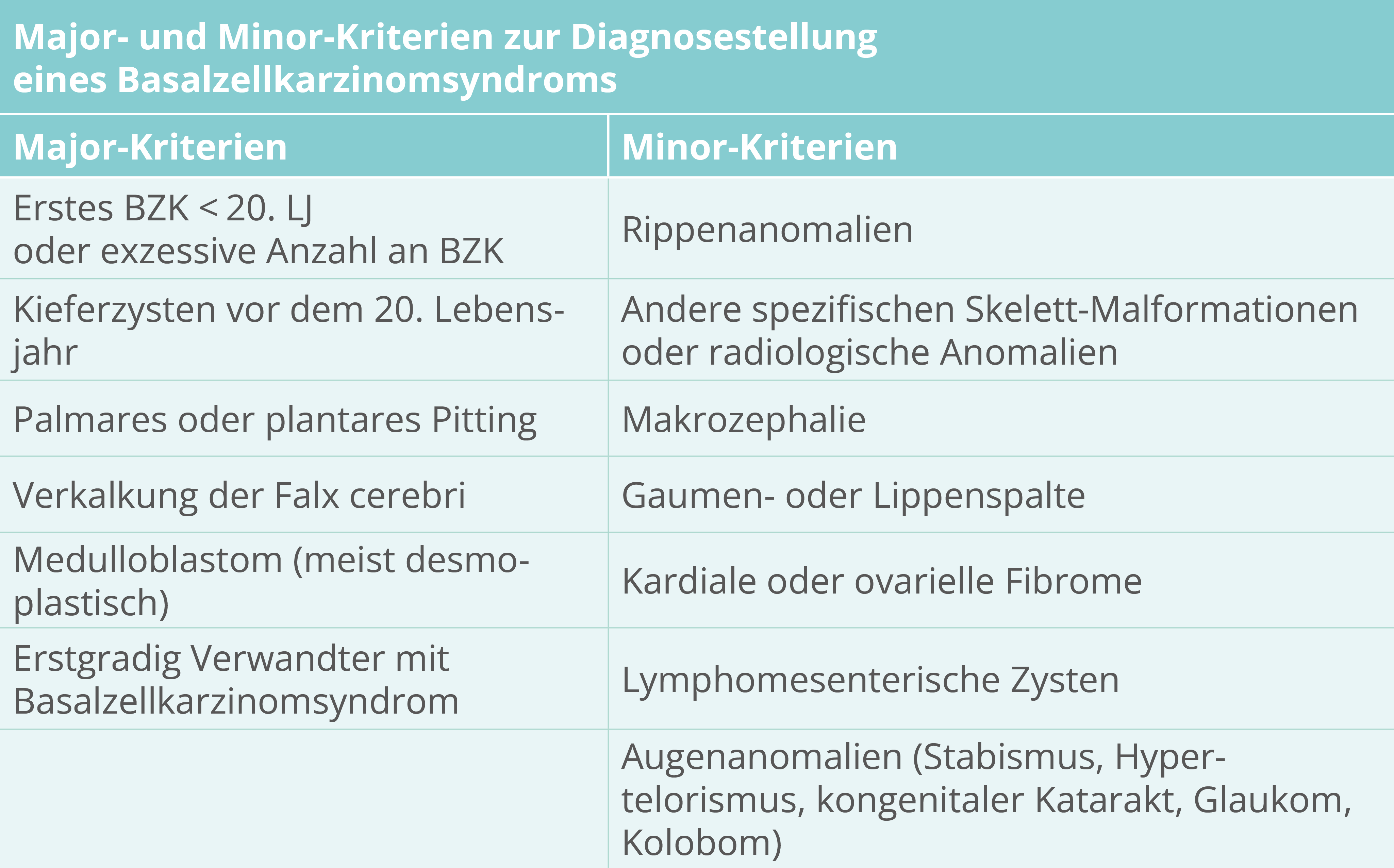
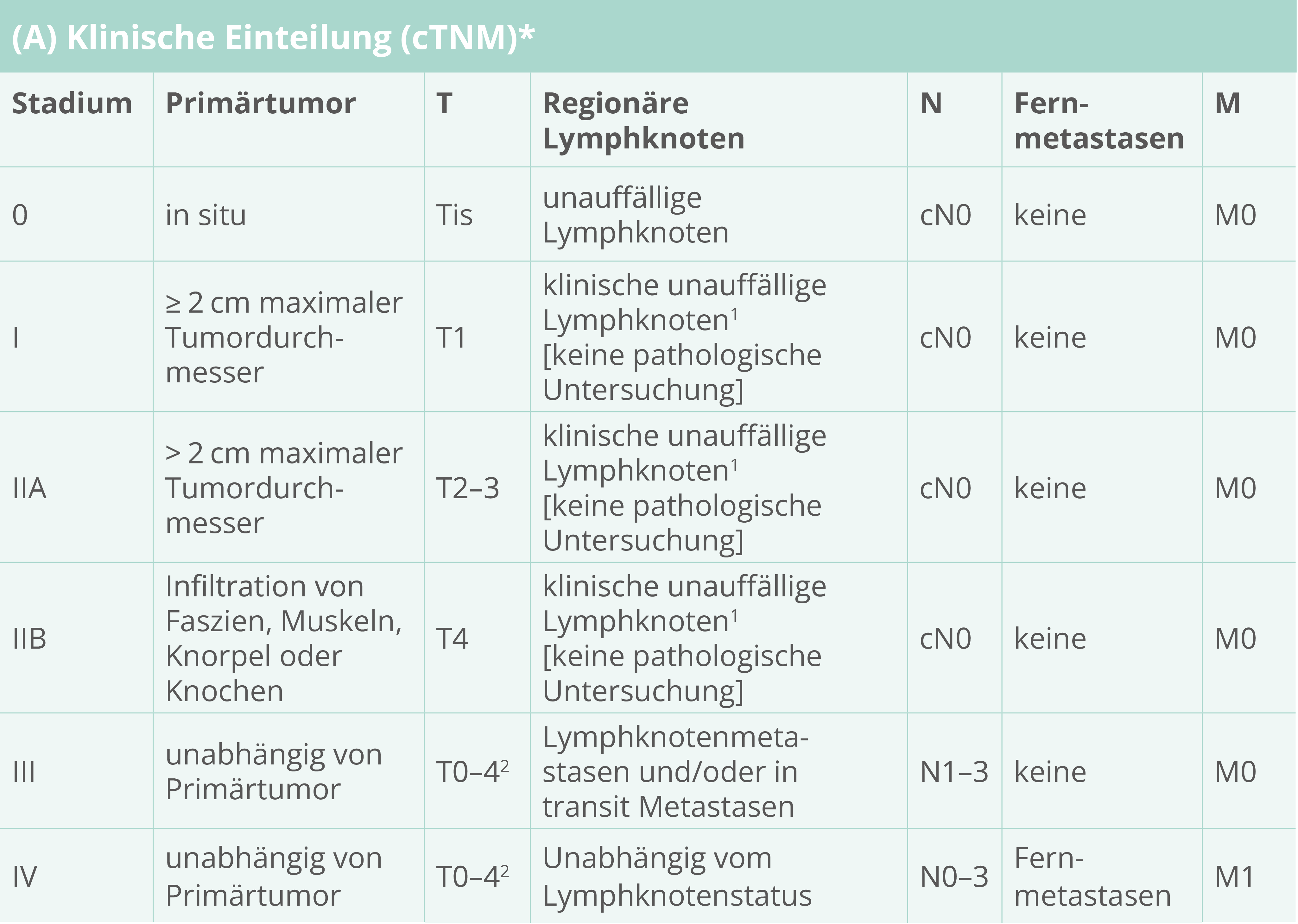
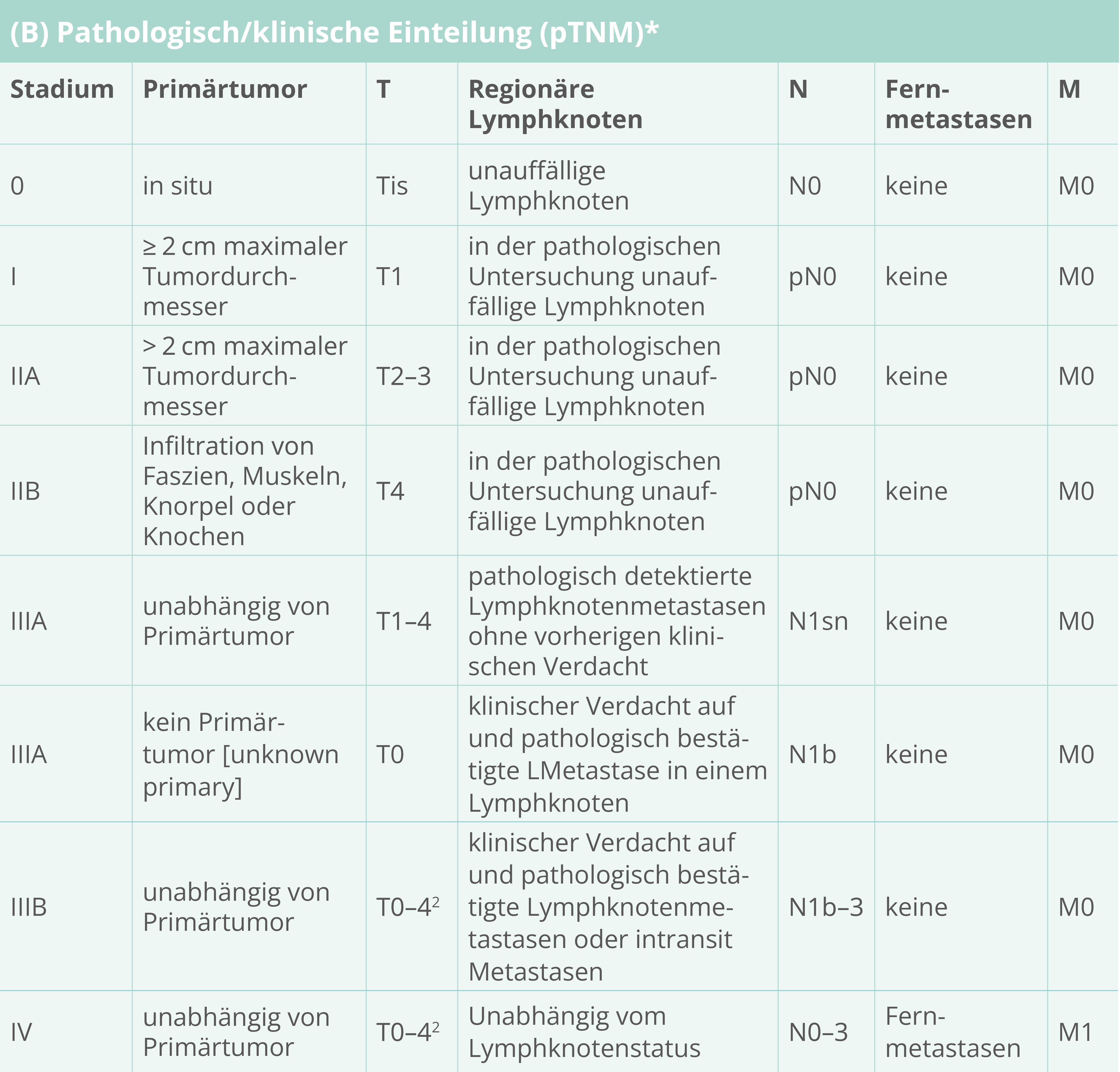
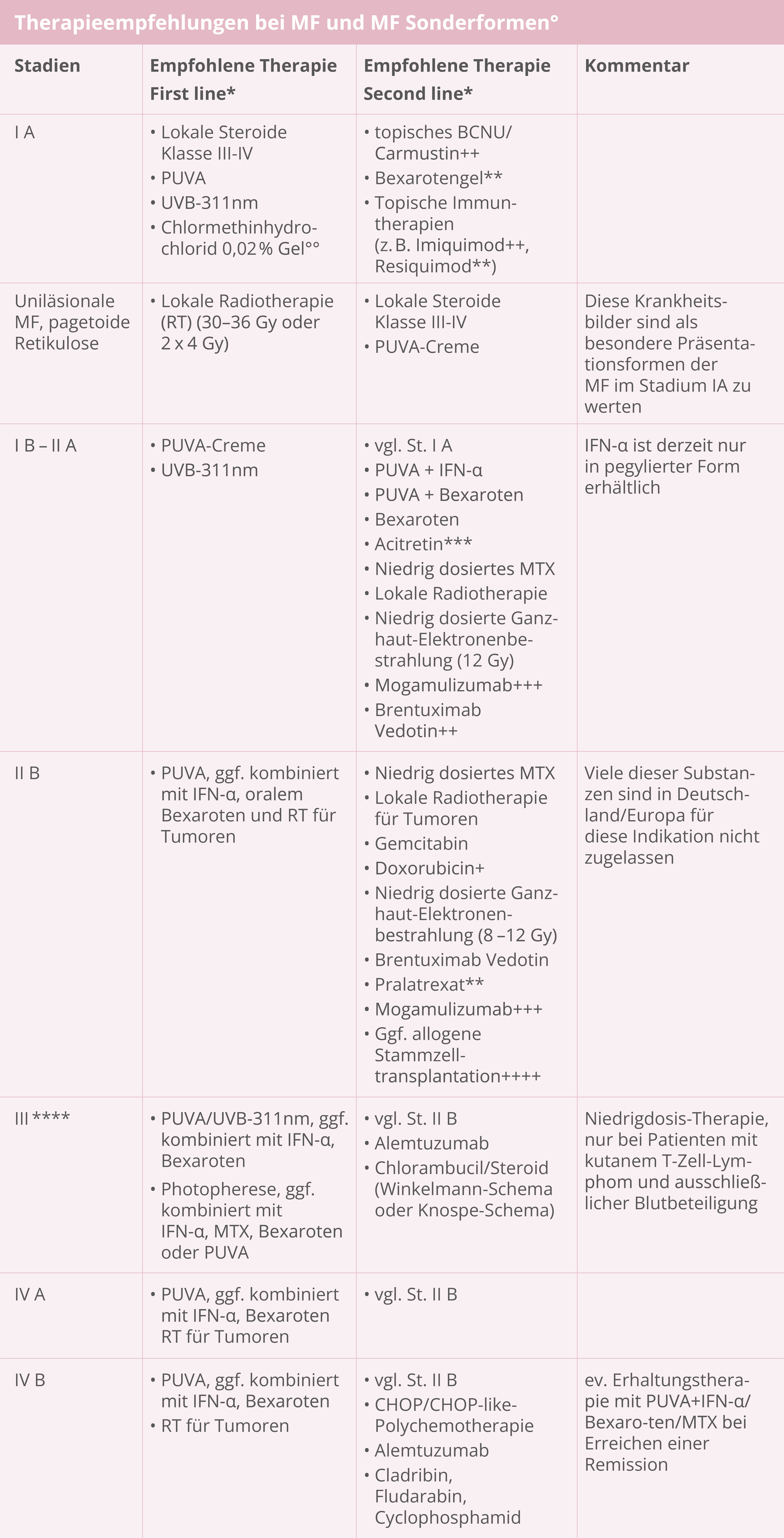
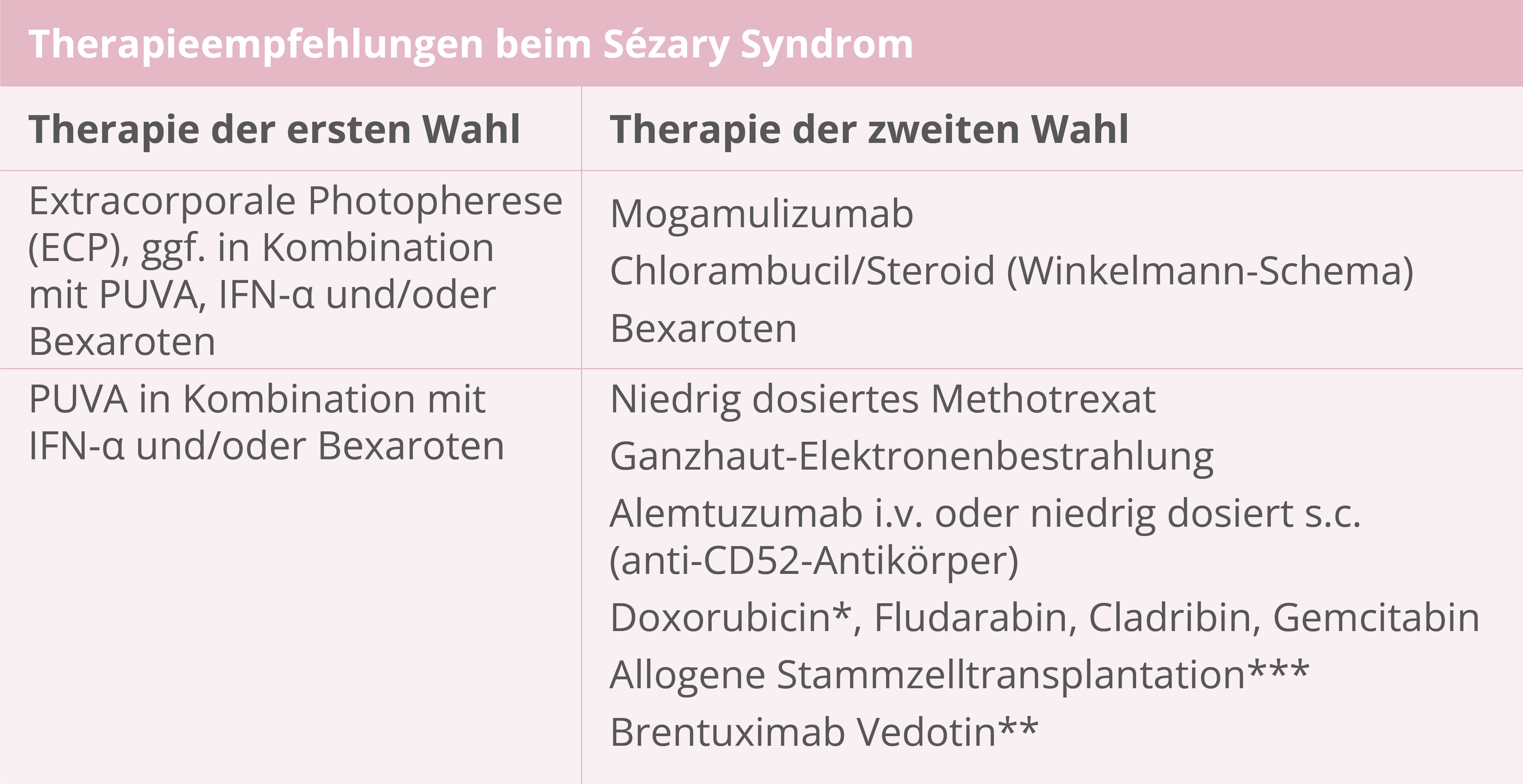
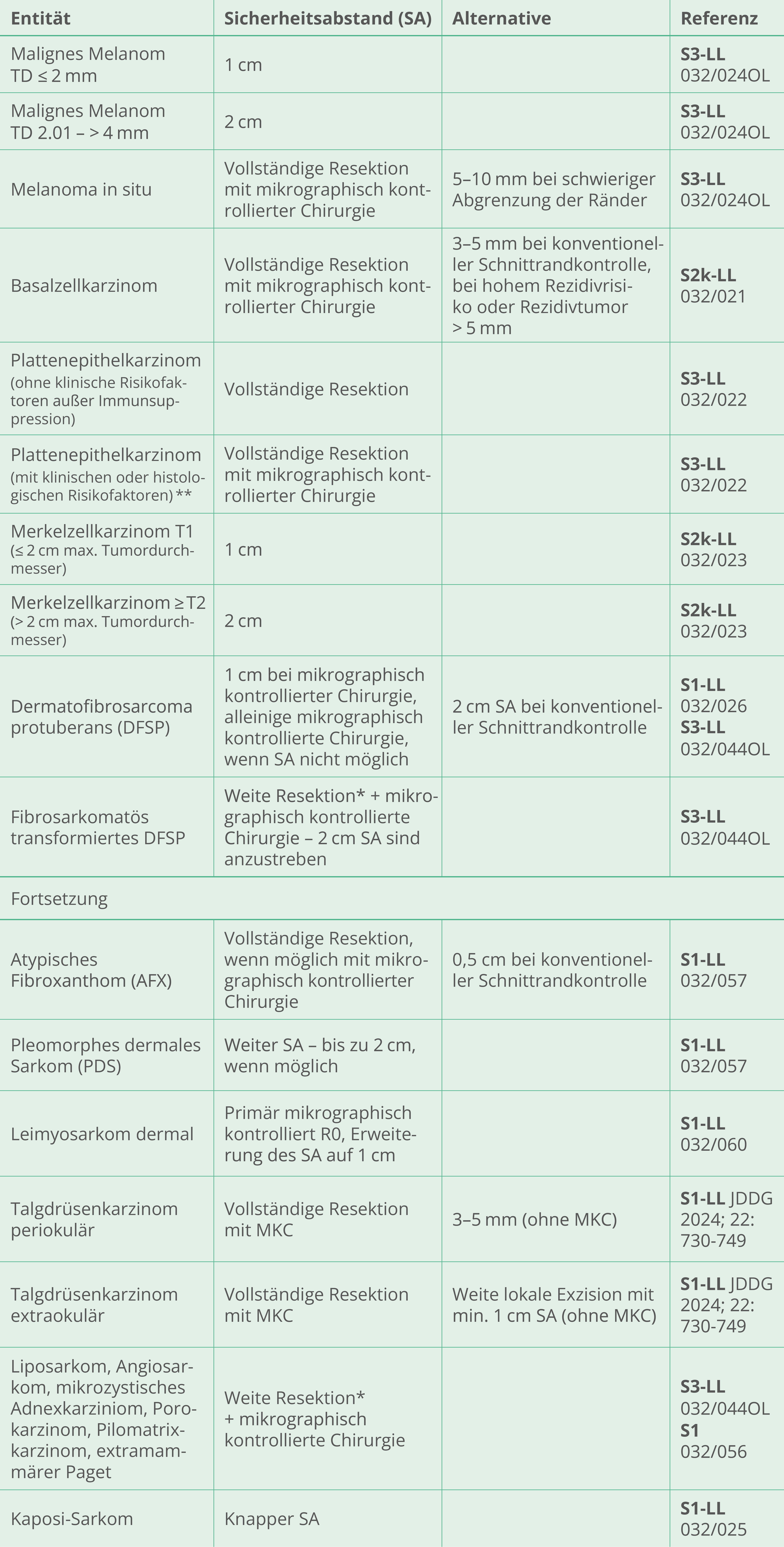
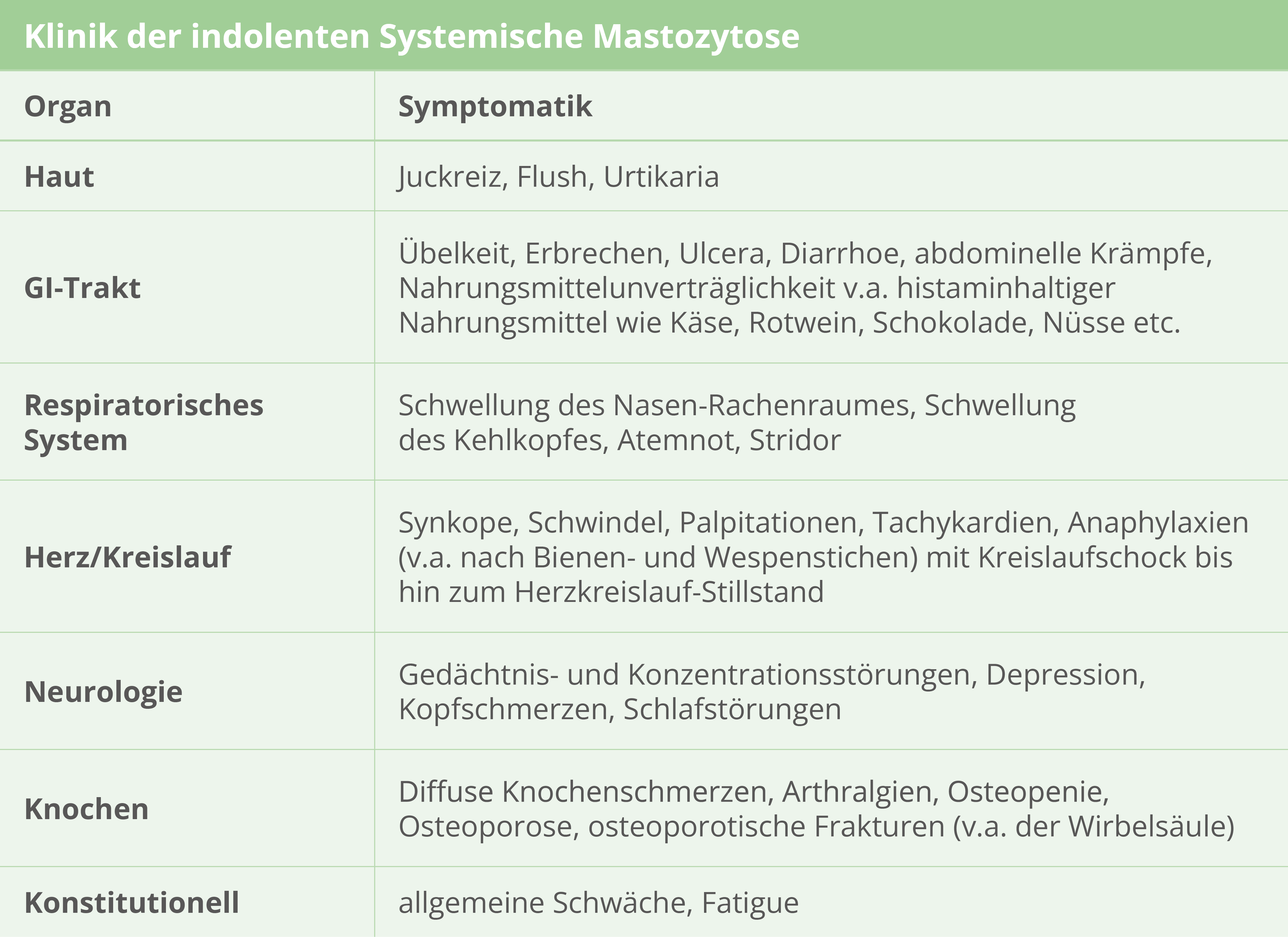
Primärtherapie
Abhängig von Größe, Ausbreitung und Lokalisation des Uveamelanoms
OP
Große Tumoren und solche mit skleraler oder extraskleraler Ausbreitung werden meist durch eine Enukleation behandelt oder mit Protonen bestrahlt.
Strahlentherapie
Kleinere Tumoren können mittels Brachytherapie oder Protonenbestrahlung behandelt werden. Strahlentherapeutische Verfahren zeigen ähnliche Abheilungsraten und haben gegenüber der Enukleation den Vorteil des Bulbuserhalts. Irismelanom: Risiken bei Strahlentherapie von Irismelanomen sind die im Verlauf mögliche Sekundärglaukombildung, Katarakt und ggf. erhöhte Blendungsempfindlichkeit. Kleinere Irismelanome können auch Augen-erhaltend reseziert werden.
Histologie und molekulargenetische Untersuchung
Diagnosesicherung: häufig kann ein Uveamelanom klinisch diagnostiziert werden, d. h. die Entnahme einer Probe zu Histologie und ggf. Molekulargenetik muss abgewogen werden, da
- Komplikationsrisiko wie z. B. Netzhautablösung, Blutungen oder Infektionen existiert.
- Molekulargenetische Untersuchungen nicht von allen Patienten gewünscht werden, z. B. Bestimmung des Chromosom 3-Status (Monosomie 3 vs. Disomie 3), Genexpressionsprofil (1). Diese Informationen sind u.a. entscheidend für die Prognose und die Planung der Nachsorge.
- Molekulargenetische Untersuchungen können ggf. im Falle einer
Medikamentöse Therapie
Eine Vorstellung in einer interdisziplinären Tumorkonferenz in einem spezialisierten Zentrum sollte zur Festlegung der Therapieoptionen erfolgen.
Klinische Studien
Für Patienten mit Uveamelanom ist die Teilnahme an klinischen Studien zu neuen Therapieansätzen eine wichtige Option.
Tebentafusp
Tebentafusp ist ein bispezifisches Fusionsprotein und die erste zugelassene systemische Therapie für HLA-A*02:01-positive Patienten mit inoperablem oder metastasiertem Uveamelanom, Verbesserung des Gesamtüberlebens in Studien.
Das Medikament wird intravenös verabreicht und erfordert eine sorgfältige Überwachung auf Nebenwirkungen, insbesondere auf das Zytokin-Freisetzungssyndrom (CRS) und Hautreaktionen.
Immuncheckpoint-Inhibitoren (ICIs)
ICIs wie Ipilimumab und Nivolumab können in Kombination oder als Einzeltherapie eingesetzt werden. Die Wirksamkeit von ICIs beim Uveamelanom ist jedoch im Vergleich zum kutanen Melanom geringer.
Andere Systemtherapien
Für Patienten, die nicht auf Tebentafusp oder ICIs ansprechen, können grundsätzlich weitere Therapieoptionen wie Chemotherapien, zielgerichtete Therapien (bislang keine Zulassung) oder zelluläre Immuntherapien überlegt werden. Ein Studieneinschluss ist hier anzustreben. Je nach Allgemeinzustand kann auch ein Vorgehen im Sinne von best supportive care sinnvoll sein.
Lebergerichtete Verfahren
Die Studienlage zu lokalen Leberverfahren ist begrenzt und die Ergebnisse sind uneinheitlich.
In einigen Studien zeigten lokale Leberverfahren eine Verbesserung des PFS und der Ansprechraten im Vergleich zur systemischen Therapie, aber keinen Vorteil im Gesamtüberleben.
Lokale Leberverfahren können bei Patienten mit metastasiertem Uveamelanom in Betracht gezogen werden, insbesondere wenn eine hohe Tumorlast in der Leber und kein oder geringer extrahepatischer Befall vorliegt.
Zu diesen Verfahren gehören:
Hepatische Chemosaturation/Chemoperfusion: Hierbei wird die Leber isoliert perfundiert, um eine hohe Konzentration an Chemotherapeutika direkt in den Tumor einzubringen.
Selektive Interne Radiotherapie (SIRT): Radioaktive Mikrosphären werden
über die Leberarterie in den Tumor injiziert.
Transarterielle Chemoembolisation (TACE): Eine Kombination aus Chemotherapeutika und Embolisationsmaterial wird in die Leberarterie injiziert, um den Tumor von der Blutversorgung abzuschneiden.
Radiofrequenzablation
Resektion: Die chirurgische Entfernung von Lebermetastasen ist nur bei isolierten Metastasen sinnvoll. Da die Leber bei Uveamelanomen oft diffus infiltriert ist, ist eine Resektion selten eine Option. Die Rezidivrate nach Resektion ist hoch.
Die Wahl des optimalen lokalen Leberverfahrens (z. B. Chemoperfusion vs. TACE vs. SIRT) hängt von verschiedenen Faktoren ab, wie der Größe und Lage der Metastasen, der Leberfunktion und dem Allgemeinzustand des Patienten.
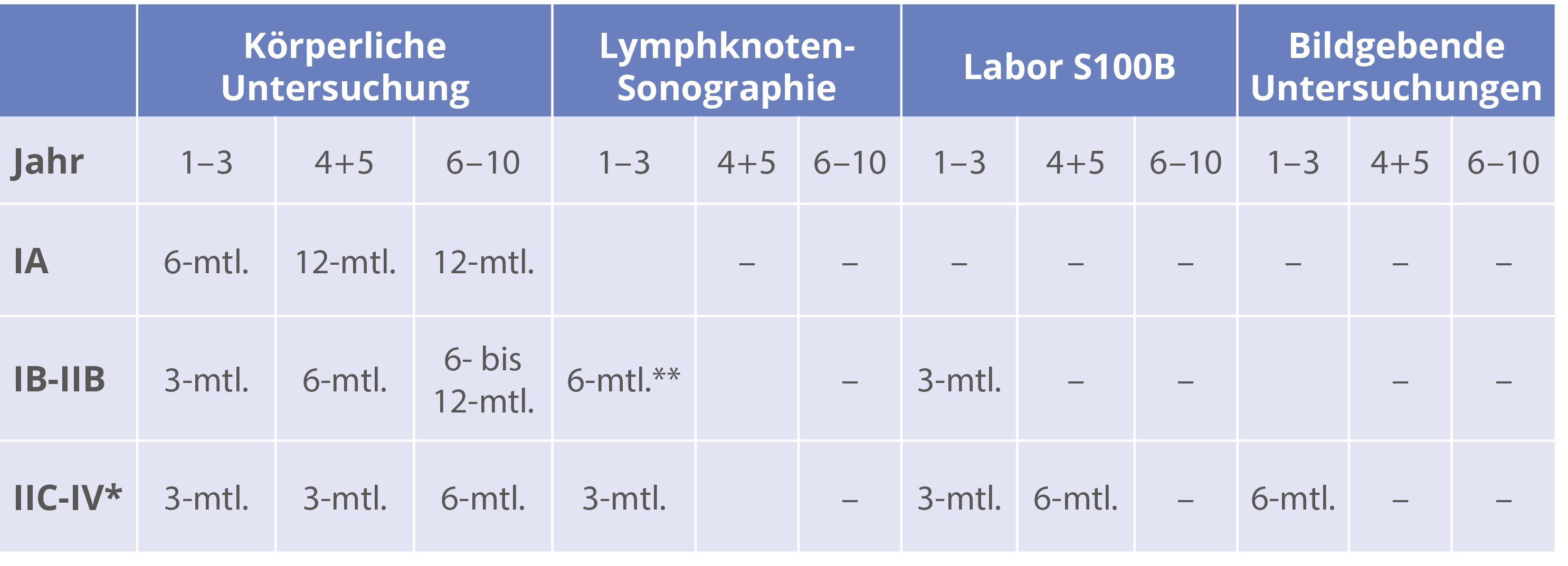
Nachsorge
Die Nachsorge umfasst:
Augenärztliche Untersuchung: z. A. Lokalrezidiv
Lebersonographie: z. A. Lebermetastasen
Labor: Transaminasen, Cholestaseparameter, LDH und S100
Bei unklaren Befunden: MRT der Leber und weitere Schnittbildgebung
Die Empfehlungen richten sich idealerweise nach dem Risikoprofil des Tumors (siehe NCCN guidelines: uveal melanoma, von 01/2024 (3):
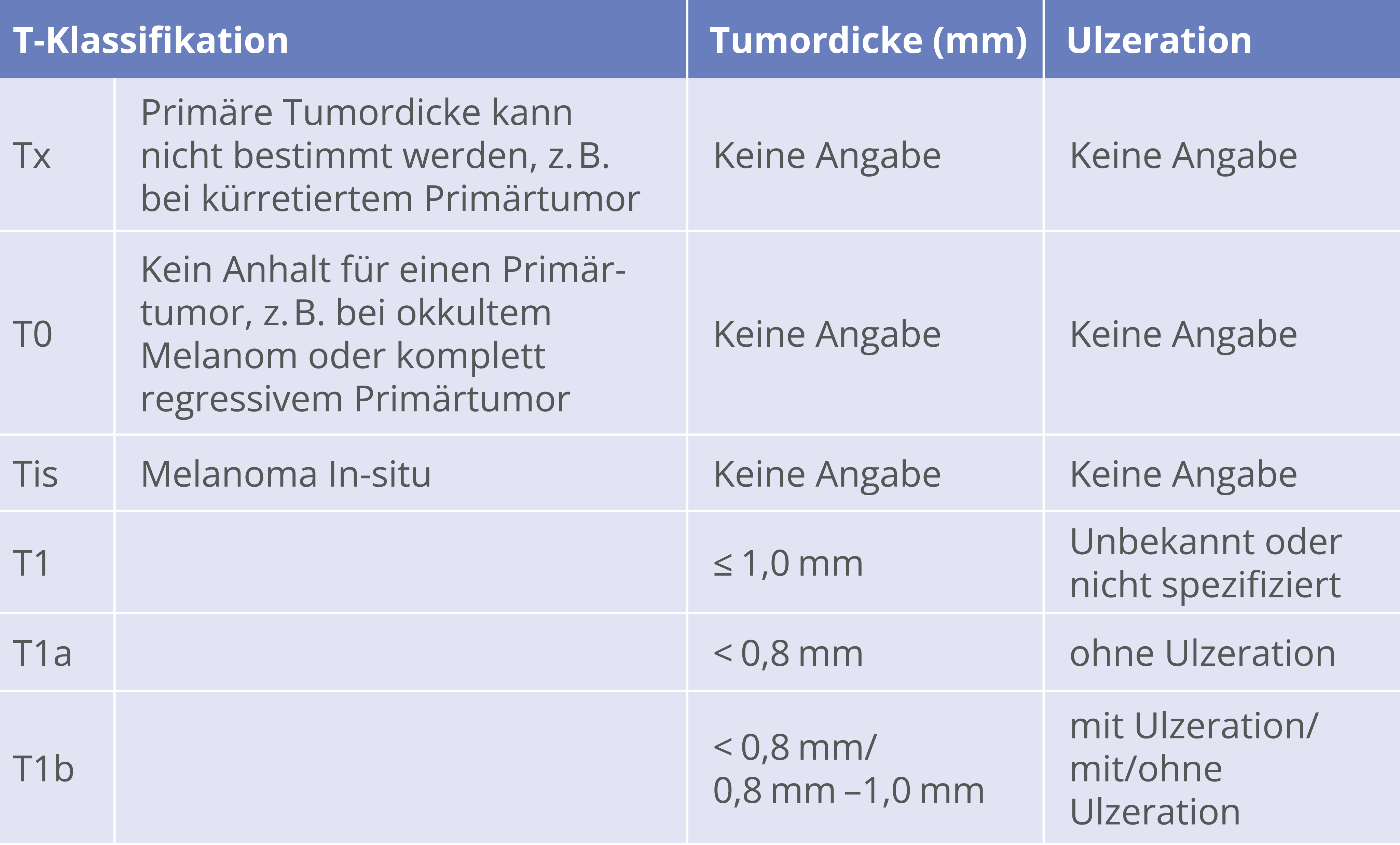
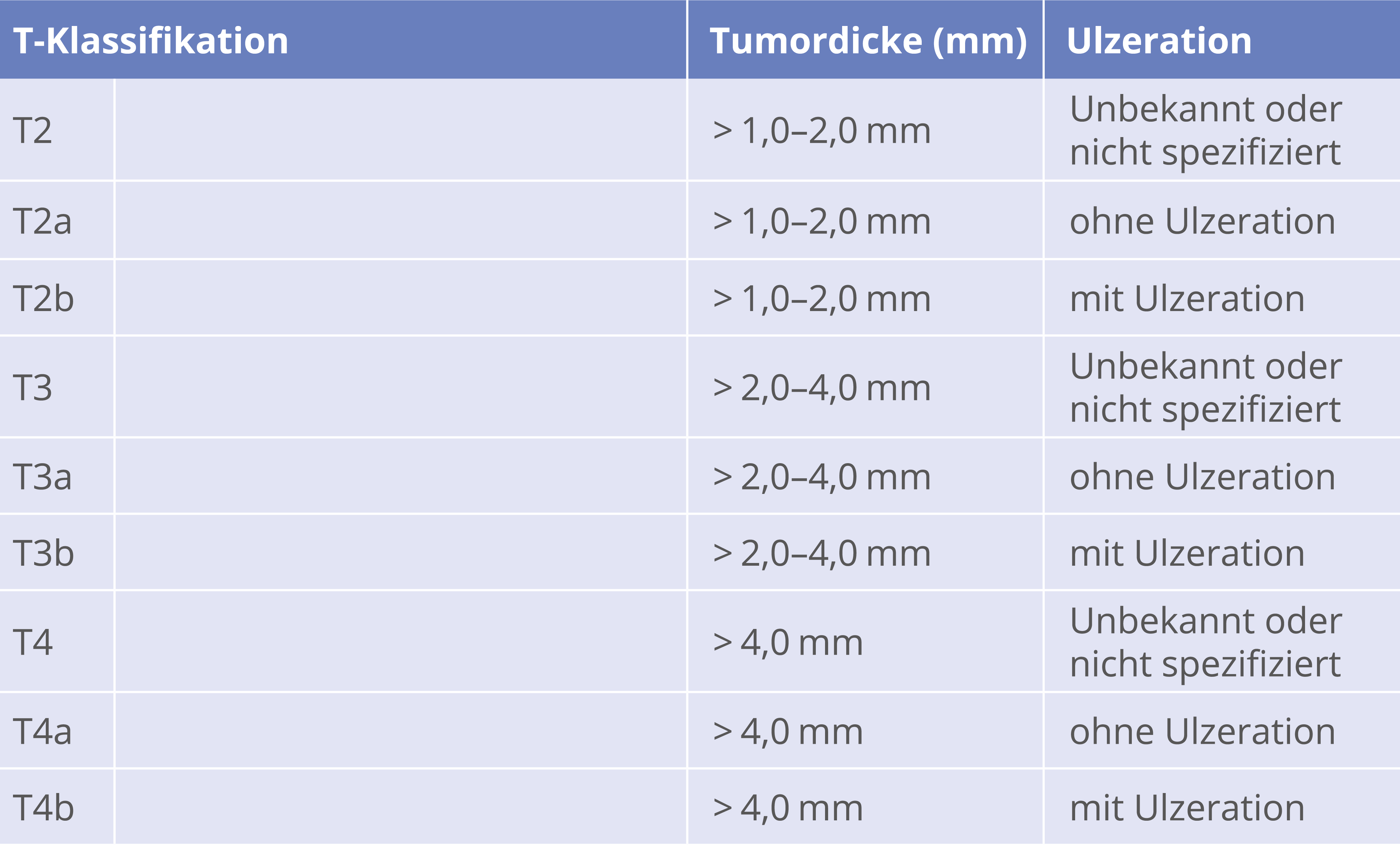
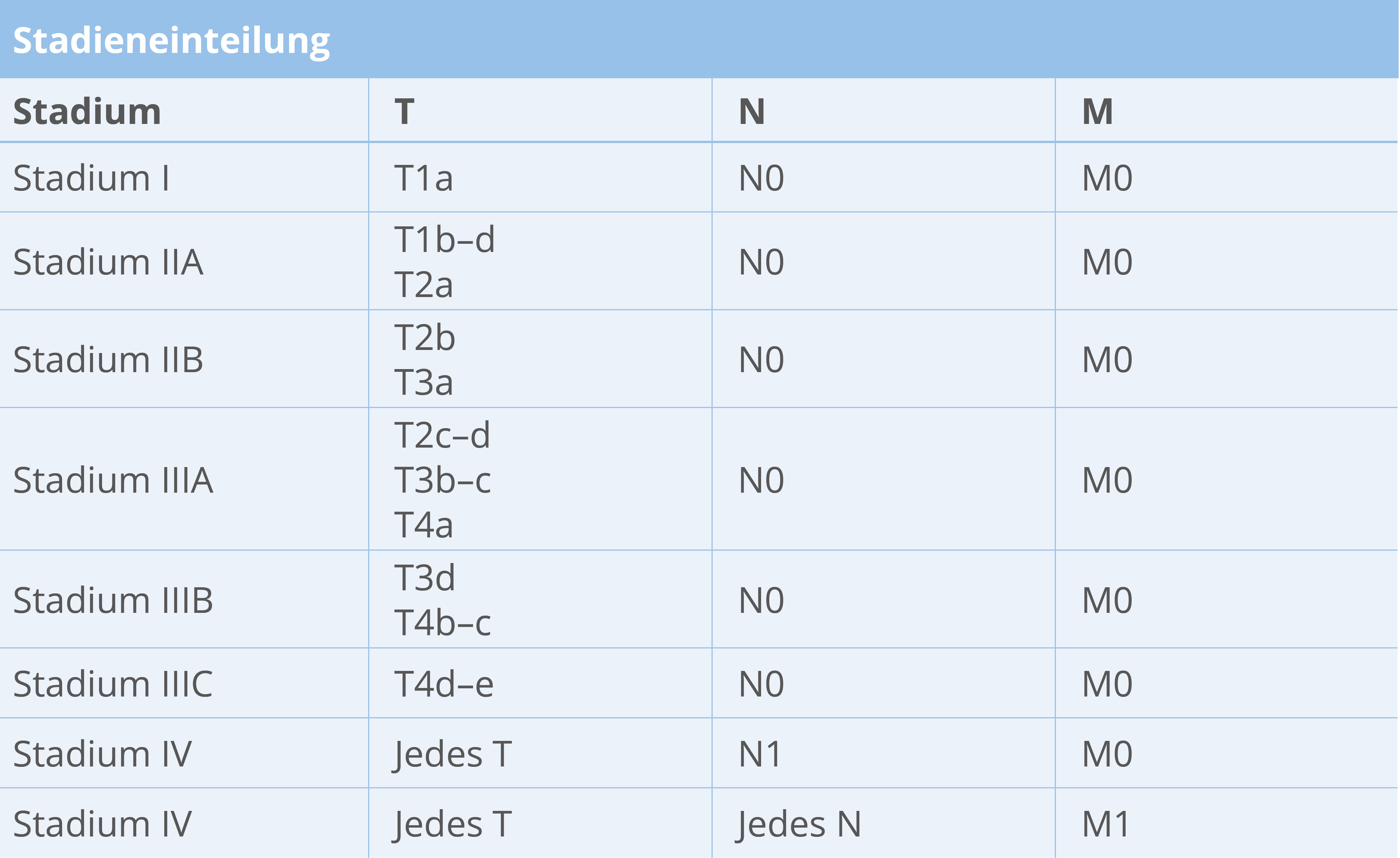
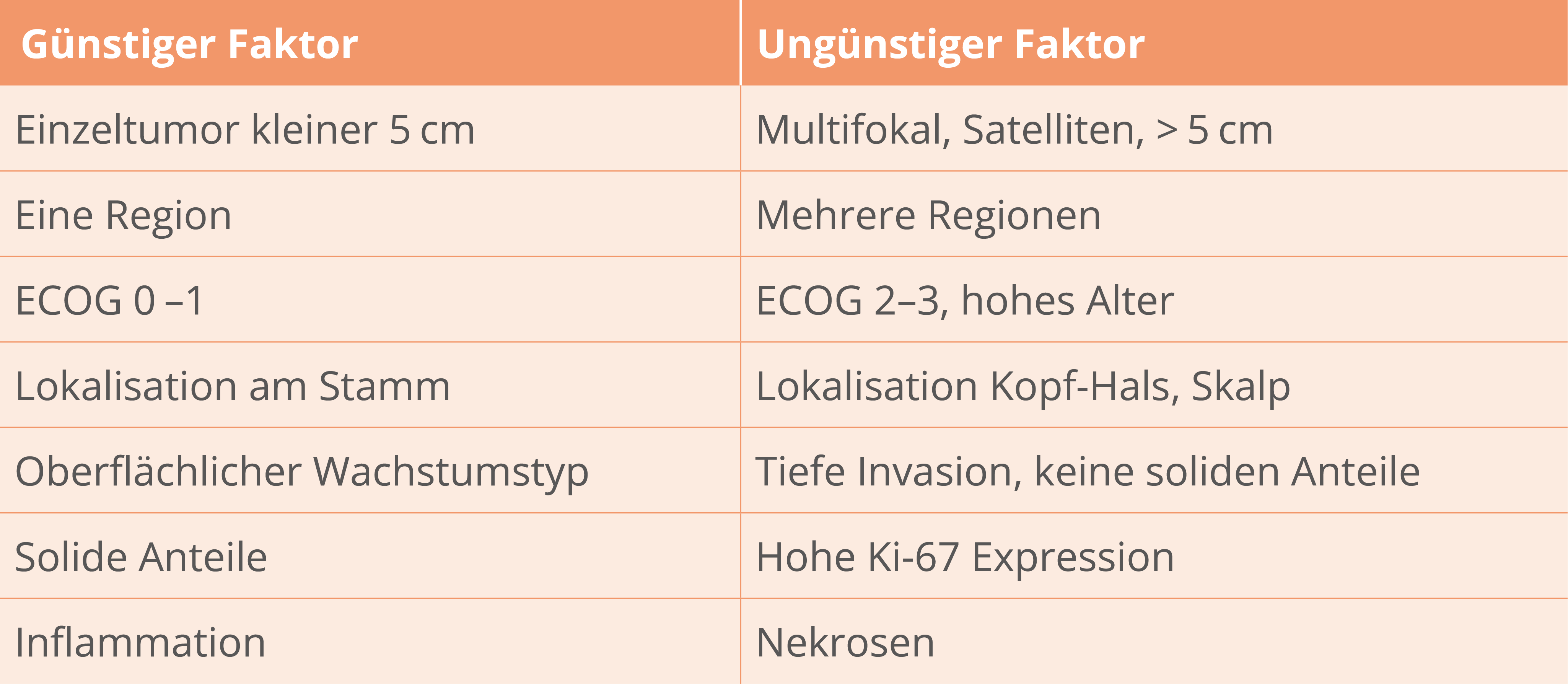
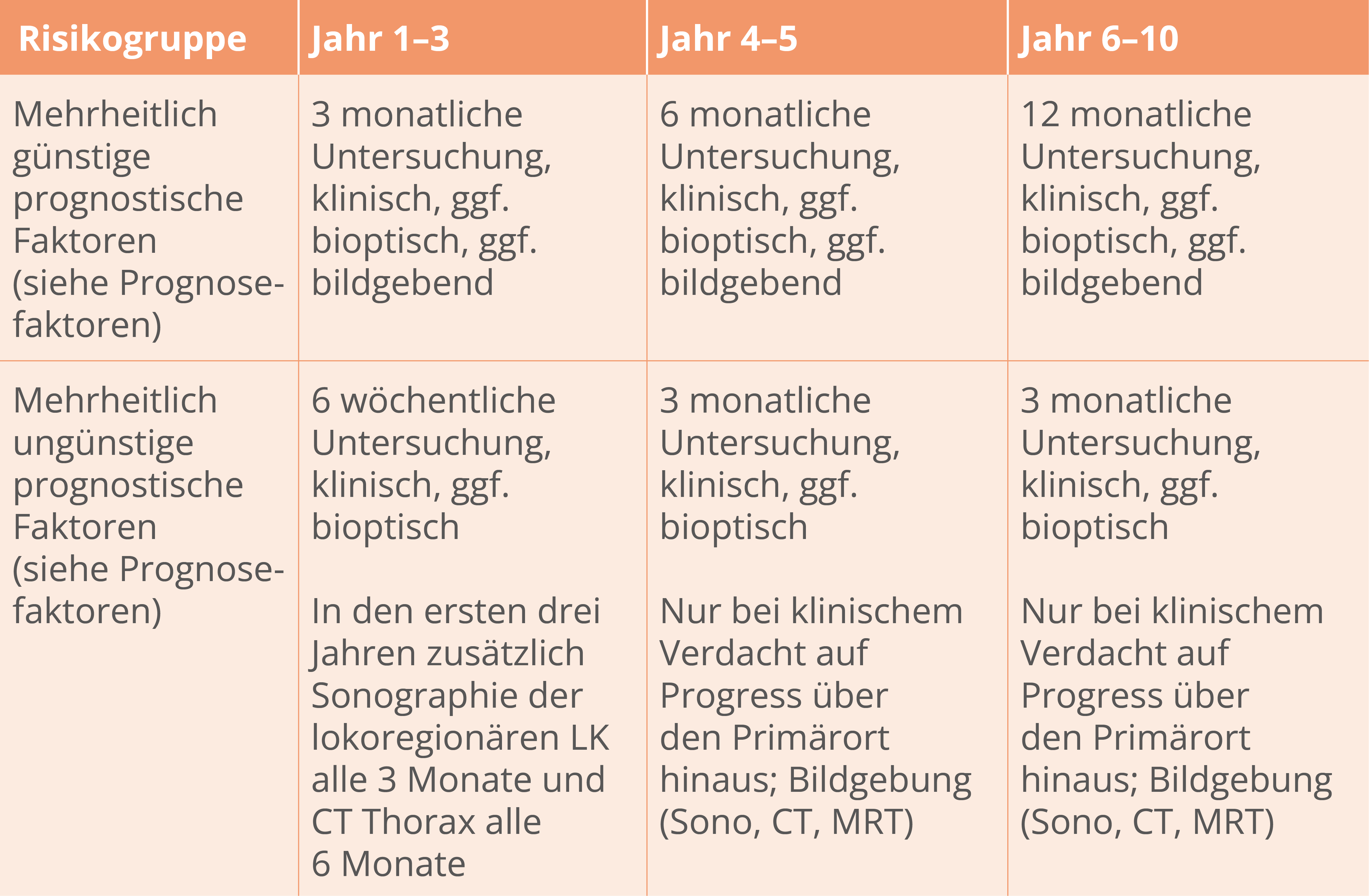
Hochrisikoprofil – T4 (AJCC) , Monosomie 3, BAP1 Mutation, Zugewinn Chromosom 8q, Genexpressionsprofil Klasse 2 nach (1): Untersuchungen alle 3–6 Monate in den ersten 5 Jahren, danach alle 6 –12 Monate bis Jahr 10, anschließend nach Klinik
Mittleres Risikoprofil – T2 und T3 (AJCC), SF3B1 Mutation, Genexpressionsprofil Klasse 1B nach (1): Untersuchungen alle 6 –12 Monate bis Jahr 10, anschließend nach Klinik
Niedriges Risiko – T1 (AJCC), Disomie 3, Zugewinn Chromosom 6p, EIF1AX Mutation, Genexpressionsprofil Klasse 1A nach (1): Untersuchungen alle 12 Monate bis Jahr 5 oder nach Klinik
Weitere Informationen
Adaptiert nach
- Onken MD, et al. Collaborative Ocular Oncology Group report number prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012;119:1596-1603.
- Hassel JC, Heppt MV: Uveamelanome, Die Onkologie 8/2023; 705-10.
- NCCN Guidelines Melanoma: Uveal; V1.2024; UM-4 ; https://www.nccn.org/login?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/uveal.pdf (abgerufen 06.01.2025)